Huntington’s Disease (HD) is a devastating neurodegenerative disorder that affects approximately one in every 20,000 Americans. HD’s namesake, George Huntingon, was not the first but the most acclaimed reporter of the disease. In 1872, when he was just 22 years old (!), he published his paper “On chorea,” in which he described the clinical features of the disease.1 Symptoms of HD include personality changes, motor impairments, and cognitive decline.

George Huntington. From Bates, 2005.
Although his report was made before the 1900 rediscovery of Gregor Mendel’s remarkable work with garden peas, Huntington recognized HD as an autosomal dominantly inherited disorder. That is, just one copy of the mutated gene will affect carriers, typically in their middle age—potentially after they have had children and passed on the gene.

Sample pedigree of a HD family, from Wikipedia.
The HD gene, or HTT, was discovered in 1983. Later, it was found that the HD-causing mutation is a “CAG-repeat expansion.” This means that carriers of the mutation have too many repeats of the nucleotides C, A, and G (in that order), which code for the amino acid glutamine. This leads to the production of a harmful HTT protein. Non-mutated HTT has 10-35 glutamines, whereas mutated HTT contains 40 or more. The number of glutamine repeats changes during transmission from one generation to another and is inversely related to the age of onset of HD (i.e., more repeats = earlier onset).2 In HD neurons, the excess glutamine leads to the aggregation of the mutant HTT (mHTT). Accumulation of mHTT also causes the formation intranuclear inclusion and apoptosis.
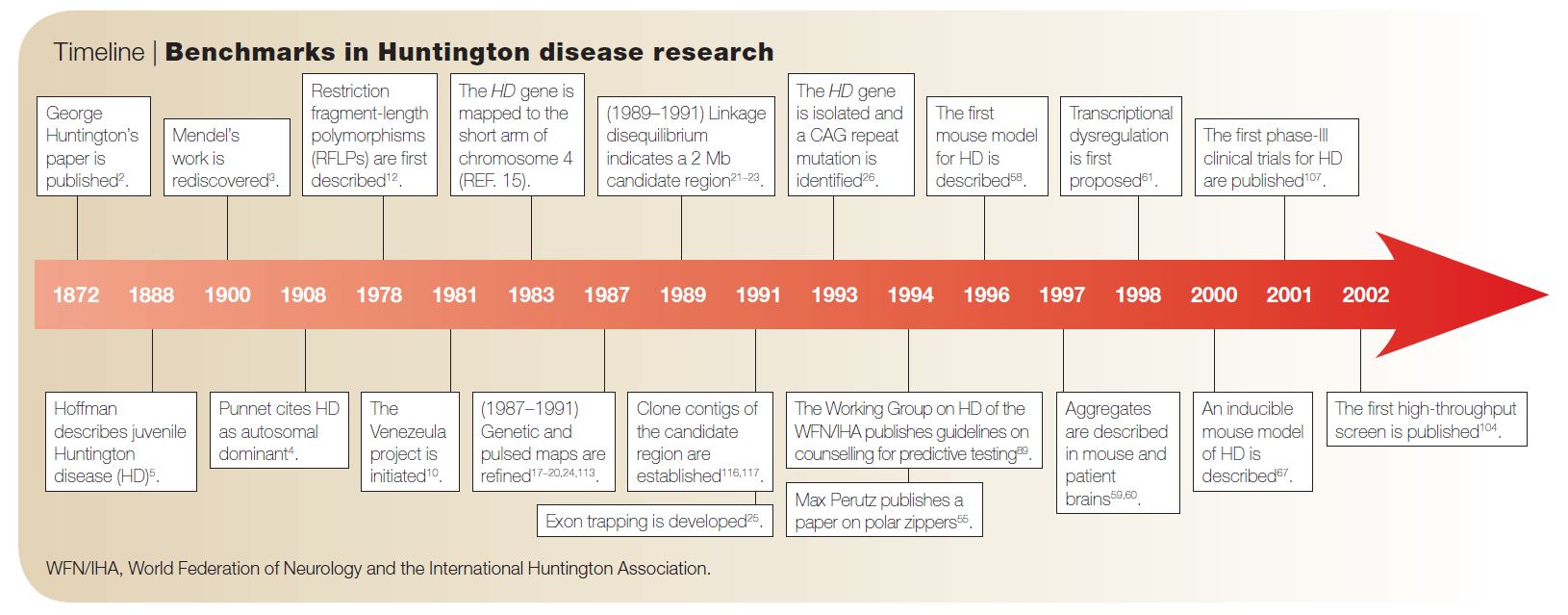
Timeline of important HD discoveries. From Bates, 2005.
Previously, it was established that the formation of excitatory cortical and striatal connections is impaired in HD brains. The mechanism behind this impairment, however, was unknown. In July of 2014, McKinstry et al. reported that Htt function is necessary for the proper development of excitatory connections in mice.3 When Htt was silenced in baby mice, cortical and striatal excitatory synapses formed and matured at an accelerated pace but then deteriorated after about 5 weeks. This same phenotype was observed in a mHtt knock-in model. Therefore, it seems that the presence of mHtt leads to loss of normal Htt function, resulting in impaired connectivity in HD brains.
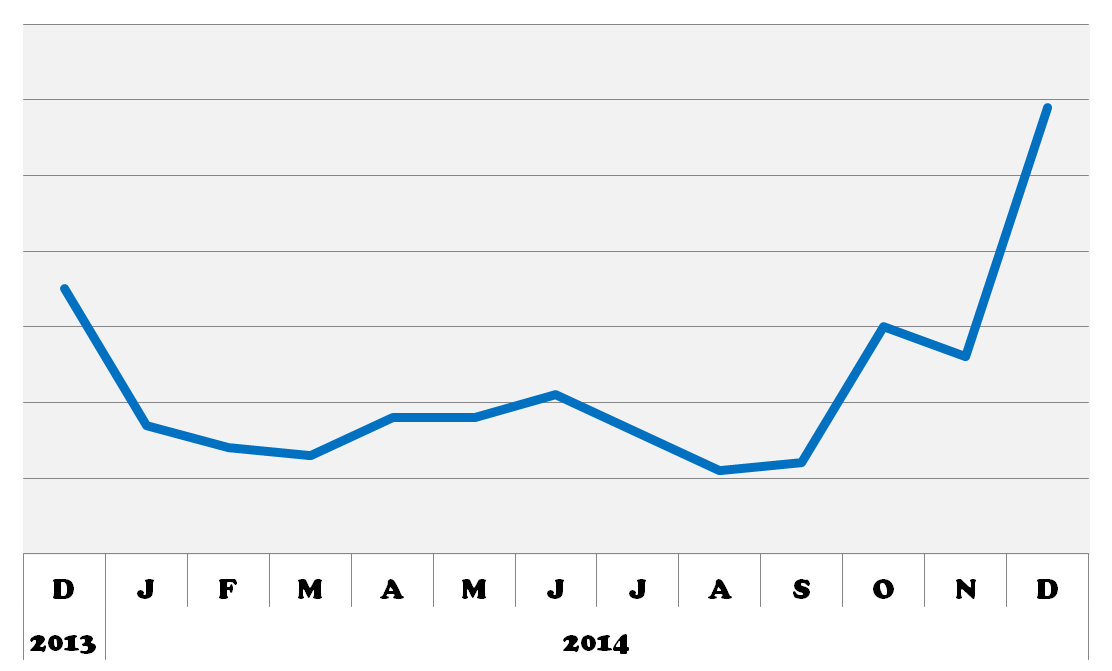
BioGPS queries for HTT. 2014 was an exciting year for HD research! I can’t wait to see what’s in store for 2015!
Also in 2014 (a big year for HD research!), Tong et al. reported that dysfunction of striatal medium spiny neurons (MSNs) may be caused by disturbances in astrocyte-mediated potassium (K+, Kir4.1) channel expression. Kir4.1 K+ channel expression is decreased in mouse models of HD, which causes elevated striatal extracellular K+. This excess extracellular K+ appears to cause increased MSN excitability. Excitingly, rescuing the loss of Kir4.1 channels via viral delivery led to the normalization of extracellular K+, improved motor function, and increased longevity in one mouse model of HD. Therefore, astrocytes and Kir4.1 channels may be good targets for HD treatment.
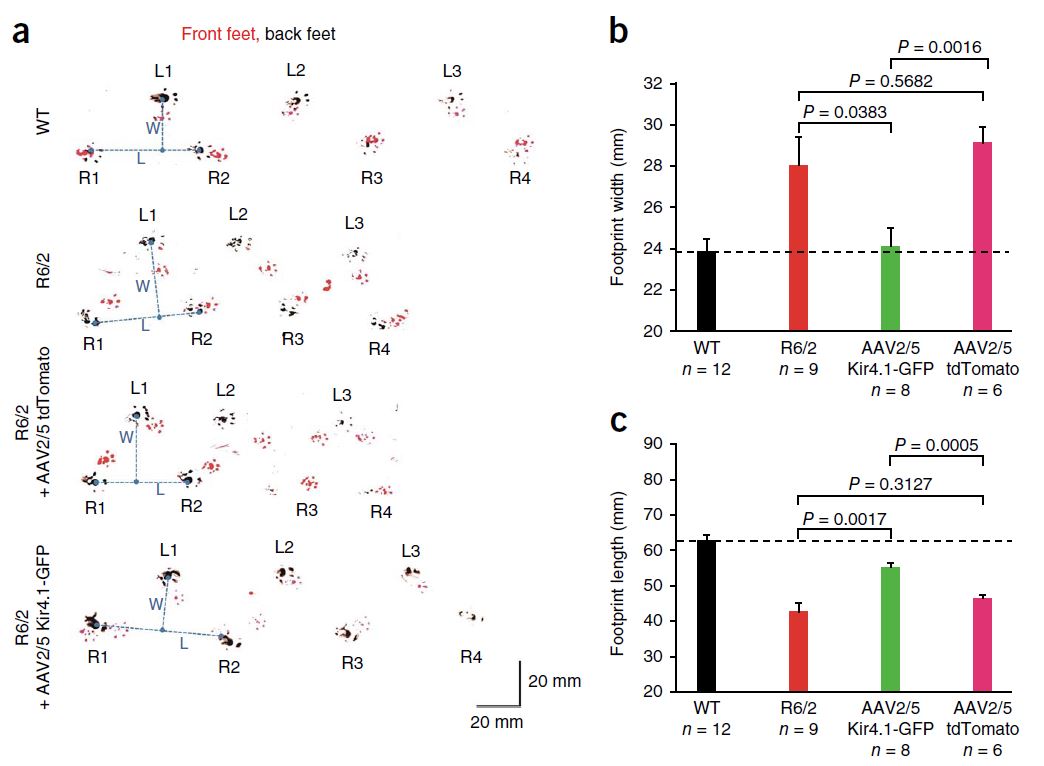
This cute assay measures mouse footprint length and width, which are too short and too long, respectively, in the R6/2 mouse model of HD. Viral delivery of Kir4.1, but not tdTomato, significantly improved this motor impairment. From Tong et al., 2014.
Speaking of HD therapies… In October of 2014, Carter et al. reported their use of induced pluripotent stem cells (iPSCs) from HD transgenic monkeys to evaluate treatments for HD.4 They found that genetic suppression of mHTT reversed mHTT aggregation, apoptosis, and sensitivity to oxidative stress. Using HD monkeys and their iPSCs will greatly advance the study of HD pathogenesis and could allow for the assessment of personalized HD treatments, including genetic correction and cell therapies.
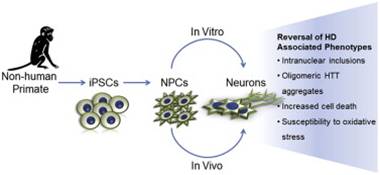
Carter et al., 2014, produced neural progenitor cells (NPCs) that matured into neurons in vivo and in vitro to model phenotypes of HD.
Meanwhile, in November, Isis Pharmaceuticals and Roche announced a phase I clinical trial for a “huntingtin lowering” therapy. This therapy, ASO-HTT-Rx, will utilize an antisense oligonucleotide (ASO) that will directly target and destroy mutant HTT messenger RNA. The ASO will be delivered to cerebrospinal fluid (CSF), much like epidurals are delivered during child birth. After an initial 4- to 6-week onset period, the gene silencing should last about 4 months. Phase I is the first phase of human trials, designed only to test safety and tolerability, not efficacy, of the drug. So there is still a long way to go before ASO-HTT-Rx will be widely available, but it’s an exciting start!5
References:
- Bates, G.P. (2005). The molecular genetics of Huntington’s disease — a history. Nat. Rev. Genet. 6, 766–773. [↩]
- Chen, S., Ferrone, F. a, and Wetzel, R. (2002). Huntington’s disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl. Acad. Sci. U. S. A. 99, 11884–11889. [↩]
- McKinstry, S.U., Karadeniz, Y.B., Worthington, A.K., Hayrapetyan, V.Y., Ozlu, M.I., Serafin-Molina, K., Risher, W.C., Ustunkaya, T., Dragatsis, I., Zeitlin, S., et al. (2014). Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J. Neurosci. 34, 9455–9472. [↩]
- Carter, R.L., Chen, Y., Kunkanjanawan, T., Xu, Y., Moran, S.P., Putkhao, K., Yang, J., Huang, A.H.C., Parnpai, R., and Chan, A.W.S. (2014). Reversal of Cellular Phenotypes in Neural Cells Derived from Huntington’s Disease Monkey-Induced Pluripotent Stem Cells. Stem Cell Reports 3, 585–593. [↩]
- Wild, E.J., and Tabrizi, S.J. (2014). Targets for future clinical trials in Huntington’s disease: what’s in the pipeline? Mov. Disord. 29, 1434–1445. [↩]